Mechanistic Study of Water Oxidation
This page was created to summarize Seth Croslow's REU experience at Boston College for the 2019 Summer
Personal Information
- Student: Seth Croslow
- Mentor: Chaochao Lang
- PI: Dr. Dunwei Wang
- College: Boston College
- Dates: June 2 - August 9, 2019
- Project Title: Mechanistic Study of Intermediates in the Water Oxidation Pathway
Background
Water oxidation is one half reaction of a larger reaction known as water splitting. Water splitting involves the breaking and rearranging of the bonds in water to form oxygen (O2) and hydrogen (H2). Water oxidation focuses specifically on the evolution of oxygen. In order to facilitate the oxygen evolution reaction (OER), catalysts are typically used. For this project, a cobalt oxide catalyst was chosen due to its abundance in the earth's crust and because it has been heavily studied within the literature as a catalyst for water oxidation. Shown below is a proposed pathway of water oxidation on a cobalt phosphate catalyst (left) and two proposed mechanisms for the oxygen oxygen bond forming step from J. Am. Chem. Soc. 2016, 138, 4229-4236.
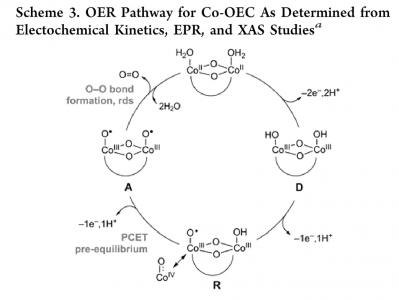
OER Pathway
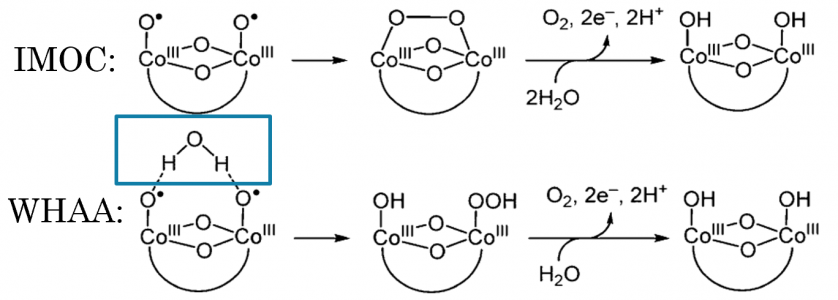
Proposed Mechanisms for Oxygen Bond Forming Step


For the two proposed mechanisms, the intramolecular oxygen coupling (IMOC) and the water hydrogen atom abstraction (WHAA), it should be noted that one key difference between the two is that the WHHA utilizes water within the mechanism. As this step is rate determining, we chose to begin the study by altering water activity to see if we can distinguish between the two mechanisms.
Electrochemistry
To begin our study, we utilized a water-in-salt method to adjust the activity of water. By adding salt into our electrolyte, the cation traps some water molecules within a solvation sheath thereby reducing the amount of water molecules available during a reaction and therefore reducing the overall activity of water. So the more salt that is added, the lower the water activity of the electrolyte solution. To estimate the overall water activity of a solution, one can calculate the mole ratio of water within the solution.
The following graph shows four water activity levels (1.00, 0.94, 0.89, 0.83) and plots them with current density on the x-axis and potential on the y-axis. The method for data collection for these trials is as follows. The catalyst was deposited onto an electrode and a CV was recorded in water activity 0. A three electrode setup was utilized including a Pt wire counter electrode, a SCE reference electrode, and the cobalt electrode as the working electrode. A linear staircase sweep voltammetry was ran which held a specific potential for 5 minutes and then increased the potential by 20 mV. The electrode was then used in the 0.94, 0.89, and 0.83 water activity solution with the same method as before. A CV was then ran again in the 1.00 water activity solution and compared to the initial CV to test the stability of the film and insure that no film degradation occurred during the trials.

Tafel Plot of Water Activity for our Cobalt Catalyst

At the lower potentials(specifically the one below 1.62 V), it is noticeable that the water activity does not change the overall current density which indicates that water is not playing a role in the mechanism. However, as the potential is increase, the current densities at each potential start to spread out with the 1.00 water activity have the largest current density and the 0.83 water activity having the lowest. This indicates that water is playing a role within the mechanism. This lead us to believe that there is a potential-dependent mechanism switch from the IMOC at low potentials to the WHAA at higher potentials. To try to validate this, we decided to use FTIR to try to look at the intermediates of the reaction and see if we can distinguish two different peaks at higher and lower potentials.
Fourier-Transform Infrared Spectroscopy
Configurations
For FTIR experiments, we began using a custom Otto Configuration that consisted of a ZnSe ATR crystal onto which we placed an FTO coated glass electrode with our cobalt catalyst face down. This, however, had large background noise and prevented evolved oxygen from escaping. We then switched to the Kretschmann Configuration which utilized a silicon ATR crystal plated with a gold film onto which our catalyst was then deposited. This configuration reduced some of the background noise and allowed oxygen to escape into the electrolyte solution.
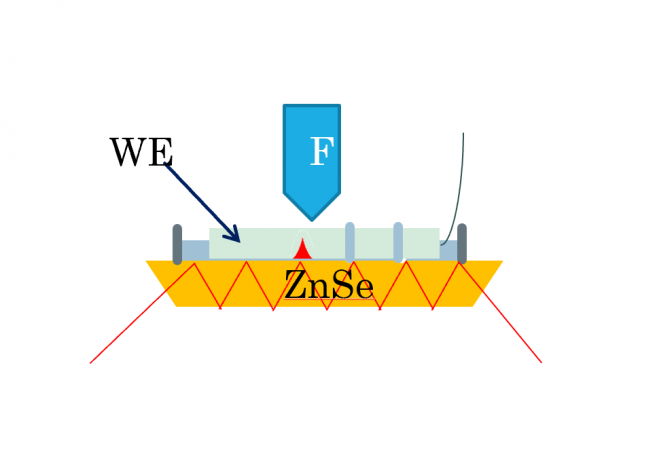
Otto Configuration

Kretchmann Configuration


Electrochemical Methods
During our FTIR data collection, we also ran various electrochemical methods to test the potential-dependance of our intermediates. One of the methods we used is shown below.

For this method, we start at a potential of 1.8 (or 1.6) which is where we notice an oxidation wave form on the CV. We hold this potential for 2.5-5 minutes to achieve a steady state. We then decrease the potential at a rate of 20 mV/s by 100 mV and hold; we follow this method until we get down to 1.0 V and then increase the potential in 200 mV increments until back up to the initial voltage. We collect FTIR data continuously throughout these trials at a rate of 1 scan every 4.611 seconds.
Isotopic Studies for Peak Detection
From previous literature (Nat. Chem., 2014, 6, 362-367) we know the expected frequency, or range, where our two main intermediates should be located: 1,013 cm-1 for the superoxide intermediate (Co-O-O-Co) and 740-920 cm-1 for the peroxide intermediate (Co-O-OH). The graphs shown below show FTIR data at each potential mentioned above (Electrochemical Methods Section). For these graphs, the x-axis shows the wavenumber, or frequency of IR light and the y-axis shows delta mOD, or absorbance. It should be noted that for these graph, the delta mOD is calculated by taking -1000 log(A/Ai) where A is the absorbance of each spectrum and Ai is the absorbance at the potential of 1.0V which we use as a reference.
The right side of the graph indicates the potential for each line starting at 1.8V at the bottom, cycling up to 1.0V in the middle, and increase back up to 1.8V at the top.
In the H2O electrolyte figure (LEFT) we can see a slight peak at 1,014 cm-1 at the bottom potential 1.8V spectra. This peak then decreases in intensity as the potential decrease and then begins to increase as the potential increase. This peak shows a potential-reversibility and indicates that it may indeed be one of our transient intermediates. However, it is hard to clearly classify this as an intermediate peak due to the large interference of the phosphate band before (~987) and after the 1,014 cm-1 peak. Additional, we see another peak at 820 cm-1 that shows up at the initial 1.8V potential, however as the potential is decreases and then increased back to the starting potential(top) the peak is no longer present which indicates that this may not be a potential-dependent intermediate.

H2O Phosphate Buffer Electrolyte
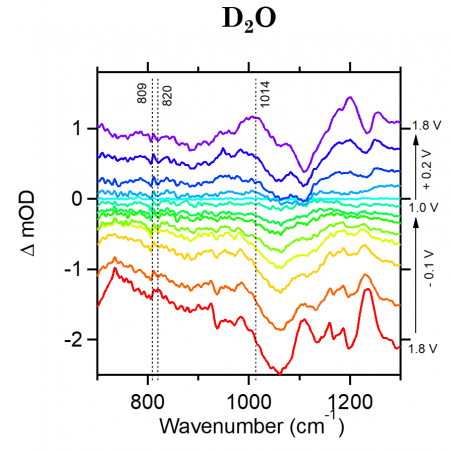
D2O Phosphate Buffer Electrolyte
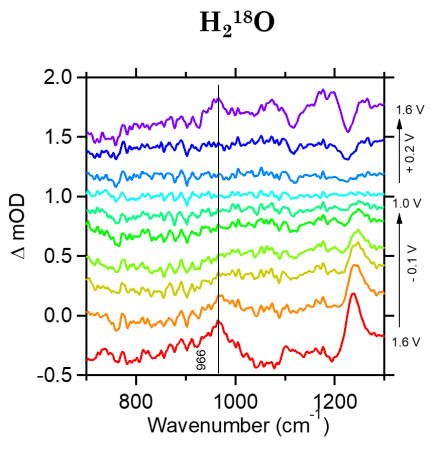
H218O Phosphate Buffer Electrolyte



Electrochemical Quartz Crystal Microbalance
Useful Articles
- Mechanistic Studies of the Oxygen Evolution Reaction by a Cobalt-Phosphate catalyst at neutral pH
- EPR evidence for co(IV) species produced during water oxidation at neutral pH
- Fe (Oxy)hydroxide oxygen Evolution Reaction Electrocatalysis Intrinsic Activity and the Roles of Electrical Conductivity, Substrate, and Dissolution
- On the chemical state of Co oxide electrocatalysts during alkaline water splitting
- Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst
- Unified structural motifs of the catalytically active state of Co(oxyhydro)oxides during the electrochemical oxygen evolution reaction