Difference between revisions of "Alex"
Jump to navigation
Jump to search
| (61 intermediate revisions by 2 users not shown) | |||
| Line 1: | Line 1: | ||
| − | Analysis of the | + | =Analysis of Phospholipid Structure through Computational Chemistry= |
| + | ==Introduction== | ||
| + | |||
| + | :Phospholipid geometry is directly influenced by the surrounding environment whether that be due to interactions with water or other molecules. It is rare that we are able to observe a phospholipid without these influences, this is where computational chemistry can be used to better understand the intramolecular interactions and overall structure of phospholipids. One such phospholipid studied is 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) which a saturated phospholipid that has been investigated due to its prevalence in cellular membranes. | ||
| + | |||
| + | ===Structure of DPPE=== | ||
| + | :DPPE is the most abundant inner leaflet phospholipid and due to this, the understanding of the molecule is important for medicine and the understanding of the cellular membrane. A saturated phospholipid, it is likely that when in the cell, there will be no kinks in the tail groups of the phospholipid. However, when alone, the structure is unknown. | ||
| + | [[File:Dppe.png|thumbnail|left]] | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | |||
| + | :Structure of DPPE | ||
| + | |||
| + | ==WebMO Calculations== | ||
| + | |||
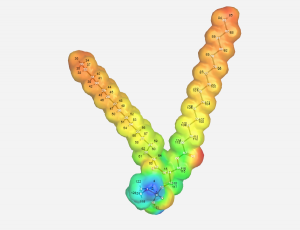
| + | :Through the use of WebMO, we are able to investigate the structure of the molecule without any of the external factors influencing the structure. This calculation was done using Hartree-Fock Gaussian (6-31G) route which gave us the best representation that was possible within the time constraints. This data set focused on geometry optimization and took 89 calculations to find the ideal structure according to the molecular energies. The overall calculation took 10:20:51. The length is due to the complexity of the molecule and the numerous intramolecular interactions that take place within the molecule. | ||
| + | [[File:Parameters.png|thumbnail|left]] | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | |||
| + | ===Results=== | ||
| + | [[File:DPPE Structure (2).gif|400px]] | ||
| + | <br /> | ||
| + | <br /> | ||
| + | ::With this file, we are able to see the various stages that the WebMO program went through in this calculation to find the lowest molecular energy which would show the most stable structure of the molecule. | ||
| + | [[File:DPPE Electron Density.png|frameless|left]] | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | <br /> | ||
| + | ::With this model of the electron density of DPPE, we can see why the tail group will push away from each other. When they are together, there will be steric hindrance due to the electrons associated with the hydrocarbon chains. | ||
| + | ===Discussion=== | ||
| + | :With this calculation, we can see that there is a spreading of the tail groups which is something that is very common in unsaturated phospholipids as there is a double bond in the hydrocarbon chain. However, in this molecule, there is no double bond as the tail groups are saturated, meaning there is no double bond to create the kink. This depiction is likely due to the intermolecular interactions that are absent in this example. As referenced in the introduction, the structure is greatly influenced by the presence of water as phospholipids have both hydrophilic and hydrophobic regions. These two areas will orient themselves in the most favorable position to exist in the environment they prefer. This likely means that the hydrophobicity of the tail groups is a much stronger force on the molecule than the steric interactions in the molecule. This force would be highly evident in experimental observations where water and other environmental factors are at play, while with computational experimentation, we can see the true steric forces acting on the molecule. | ||
Latest revision as of 21:25, 15 April 2021
Analysis of Phospholipid Structure through Computational Chemistry
Introduction
- Phospholipid geometry is directly influenced by the surrounding environment whether that be due to interactions with water or other molecules. It is rare that we are able to observe a phospholipid without these influences, this is where computational chemistry can be used to better understand the intramolecular interactions and overall structure of phospholipids. One such phospholipid studied is 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) which a saturated phospholipid that has been investigated due to its prevalence in cellular membranes.
Structure of DPPE
- DPPE is the most abundant inner leaflet phospholipid and due to this, the understanding of the molecule is important for medicine and the understanding of the cellular membrane. A saturated phospholipid, it is likely that when in the cell, there will be no kinks in the tail groups of the phospholipid. However, when alone, the structure is unknown.

- Structure of DPPE
WebMO Calculations
- Through the use of WebMO, we are able to investigate the structure of the molecule without any of the external factors influencing the structure. This calculation was done using Hartree-Fock Gaussian (6-31G) route which gave us the best representation that was possible within the time constraints. This data set focused on geometry optimization and took 89 calculations to find the ideal structure according to the molecular energies. The overall calculation took 10:20:51. The length is due to the complexity of the molecule and the numerous intramolecular interactions that take place within the molecule.

Results
.gif)
- With this file, we are able to see the various stages that the WebMO program went through in this calculation to find the lowest molecular energy which would show the most stable structure of the molecule.

- With this model of the electron density of DPPE, we can see why the tail group will push away from each other. When they are together, there will be steric hindrance due to the electrons associated with the hydrocarbon chains.
Discussion
- With this calculation, we can see that there is a spreading of the tail groups which is something that is very common in unsaturated phospholipids as there is a double bond in the hydrocarbon chain. However, in this molecule, there is no double bond as the tail groups are saturated, meaning there is no double bond to create the kink. This depiction is likely due to the intermolecular interactions that are absent in this example. As referenced in the introduction, the structure is greatly influenced by the presence of water as phospholipids have both hydrophilic and hydrophobic regions. These two areas will orient themselves in the most favorable position to exist in the environment they prefer. This likely means that the hydrophobicity of the tail groups is a much stronger force on the molecule than the steric interactions in the molecule. This force would be highly evident in experimental observations where water and other environmental factors are at play, while with computational experimentation, we can see the true steric forces acting on the molecule.